2.3.1 Optisk emissionsspektrometri med gnistexcitering
OES med gnistexcitering är utan jämförelse den vanligaste spektrometriska metoden för rutinmässig analys av stål och metaller. Anledningarna är flera: metoden är snabb, max 2 minuter per prov; hög precision och låga detekterbarhetsgränser för flertalet element; simultanmätning av så gott som alla element av intresse, idag till och med kväve. En begränsning är dock haltområdena, som är snävare än till exempel ICP och XRF.
En optisk emissionsspektrometer kan indelas i följande enheter:
- energikälla (högspänningsaggregat)
- dispersivt medium (själva spektrometern)
- utvärderingsenhet (mätelektronik och dator)
Energikällan
För att direkt kunna alstra strålning från fasta prov, måste man tillföra så hög energi att en representativ del av provet smälter och förångas. Detta kan man åstadkomma med en elektrisk urladdning. Tidigare, under 1940- och 1950-tal, använde man likströmsbåge, som ger en temperatur av 4000-5000ºC. I vissa fall används fortfarande likströmsbåge, som ger hög känslighet vid spårämnesbestämning. Problemet är dock dålig stabilitet, vilket resulterar i dålig reproducerbarhet.
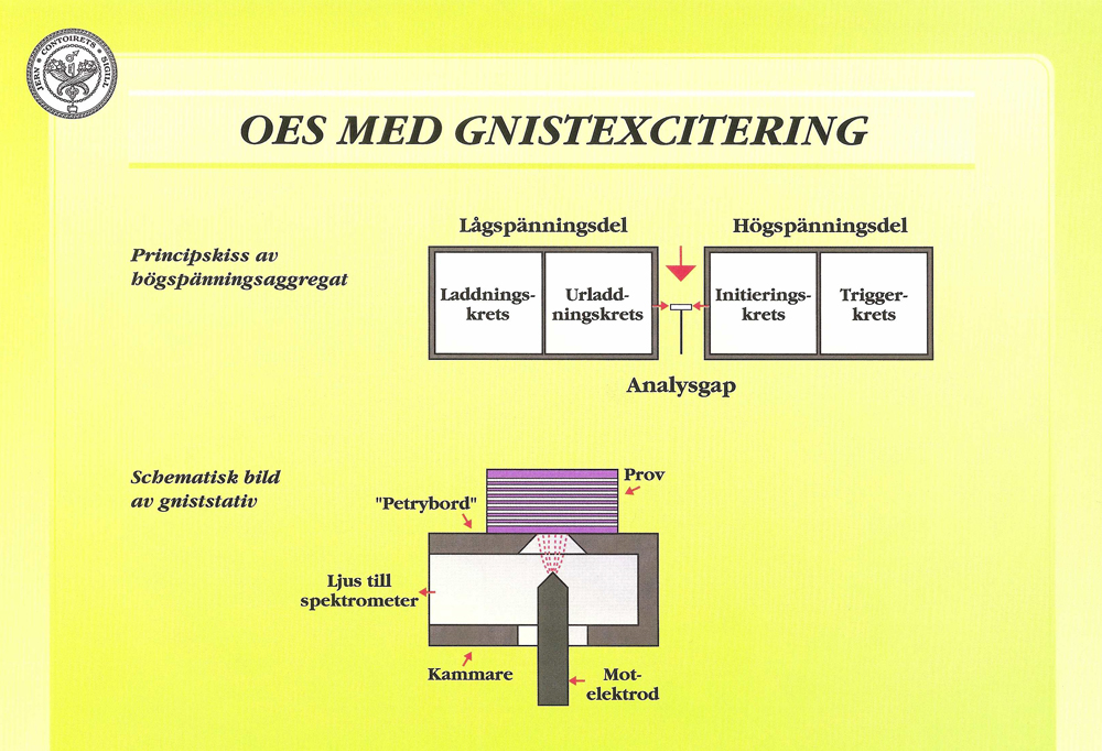
God reproducerbarhet erhålls däremot av en kondenserad gnisturladdning genererad av ett högspänningsaggregat. Figur 25:a visar schematiskt hur ett sådant aggregat är uppbyggt.
I lågspänningsdelens laddningskrets sker upptransformering och likriktning av växelspänningen samt laddning av en kondensator. I urladdningskretsen frigörs den uppladdade energin till analysgapet via motstånd och induktionsspole, vilkas värden går att variera.

Gnisturladdningen kan på så sätt påverkas så att den får en mer bågliknande karaktär. Frekvensen hos gnistan kan variera mellan 50 och 400 Hz. Då spänningen över analysgapet endast rör sig om 400 – 600 V, erfordras en initiering av gnistan. Detta sker genom högspänningsdelen, som genererar högspända gnistor med försumbar energi.
Den momentana strömstyrkan i gnisturladdningen kan nå höga värden, vilket ger temperaturer upp till 10 000 K. Detta innebär att de atomer från provet som finns i urladdningszonen inte endast exciteras i sitt grundtillstånd utan även från högre jonisationsstadier. Gnistspektrum innehåller därför betydligt fler linjer än ett bågspektrum.
Avgnistningen av ett prov brukar delas in i förgnistning och integrering. För att utjämna eventuella variationer i ytan av provet, är det vanligt att låta förgnistningen få högre energi (”HEPS”: high energy pre spark) än den efterföljande integreringen. Med modern och snabb elektronik är det nu också möjligt att diskriminera varje enskild gnista, helt eller delvis, för att förbättra analysprestanda. Moderna spektrometrar är utrustade med så kallat tidsupplöst elektronisk integrering. Detta innebär att från varje individuell gnista används endast den ”bästa” delen av signalen. Denna teknik ger lägre bakgrund samt bättre känslighet och precision.
Spektrometern
För att kunna identifiera och mäta det ljus atomerna utsänder, måste det ledas in i en spektrometer, där det delas upp i olika våglängder. I princip kan samma typ av spektrometer användas för olika ljuskällor. Två principiellt skilda spektrometrar förekommer, nämligen sekvensiella och parallella (simultana). Med den sekvensiella spektrometern mäts ett element (linje) i taget, medan den parallella mäter så många element samtidigt som den är utrustad för (vanligen upp till 60 st).
Stativet är den enhet på spektrometern där provet sätts fast. För att skydda provytan mot oxidation sker gnistningen i argon, som får strömma genom gnistkammaren, se Figur 25:b. Motelektroden är vanligen av volfram och har konisk spets. Elektrodavståndet kan variera mellan 3 och 5 mm. Vissa spektrometertillverkare utnyttjar optiska ljusledare, som kan riktas mot en viss del av gnistplasmat och leda in ljuset i en separat enhet av spektrometern.
Spektrometerns optiska del (polykromatorn) består av följande komponenter:
- kondensorlins
- ingångsspalt
- reflexionsgitter
- utgångsspalter
- speglar
- detektorer
Spektrometerns optiska utförande utgår från en så kalladRowlandcirkel, vilket innebär att ingångsspalt, gitter och utgångsspalter är placerade på en cirkel med en diameter som är lika med gittrets radie (”Paschen-Runge”-uppsättning). Detta mått benämns spektrometerns fokallängd. Vanliga fokallängder är 0,5, 0,75 och 1,0 m (större fokallängd = bättre upplösning).
Linsen samlar och fokuserar ljuset på ingångsspalten, som släpper in lämplig del av ljuset i spektrometern. Ingångsspaltens öppning påverkar i viss mån spektrometerns upplösningsförmåga. En vanlig spaltöppning är 20 μm.
Ingångsspaltens läge i fokalplanet är justerbart och bestämmer hur spektrat skall avbildas på utgångsspalterna. Justeringen kallas profilering.
Gittret spjälkar upp ljuset i sina spektrala beståndsdelar, som fokuseras och avbildas på utgångsspalterna. Öppningen på dessa avgör hur mycket ljus av respektive våglängd som skall släppas igenom till fotomultiplikatorerna. Utgångsspalterna brukar ha en öppning mellan 35 och 75 μm. Figur 26:a och Figur 26:b visar i princip strålgången och de optiska komponenternas placering. Avlänkningen av ljuset i speglarna är till för att få plats med så många kanaler som möjligt.

Gittret
Hjärtat i en optisk spektrometer är gittret. Dess förmåga att sprida ljuset benämns dispersion och är beroende av hur många ritsar gittret har per millimeter. Ju fler ritsar desto bättre dispersion. Vanliga tal är 1000-2400 linjer per millimeter för mekaniskt ritsade gitter.
Ett annat begrepp är upplösningsförmåga, vilket brukar definieras som den våglängdsseparation som gör det möjligt att skilja två linjer från varandra. Detta i sin tur är avhängigt totala antalet linjer som är belysta på gittret. Större gitter ger således bättre upplösning. En hög upplösning gör dock spektrometerns våglängdsområde mer begränsat. Vid dimensionering av en spektrometer är det således en balansgång mellan att täcka in sitt analysbehov och att samtidigt få så bra prestanda som möjligt.
Tidigare användes prisma i stället för gitter som dispersivt medium, men i dag är det så gott som uteslutande gitter som gäller. Gittret har i motsats till prismat fördelen att ha nästan linjär dispersion över hela våglängdsområdet. Dessutom kan spektrallinjer av högre ordning användas om gittret är framställt genom mekanisk ritsning. Gitter framställda med laser (holografiska) är inte lika bra för högre ordningar. Å andra sidan kan sådana gitter framställas med större linjetäthet.
Våglängder under cirka 200 nm absorberas starkt av luftens syre, varför spektrometern endera måste hållas under vakuum eller spolas med argon eller kväve. Vanliga element med linjer i detta område är kol, fosfor, svavel, bor och kväve.
Detektorer, registrering
Ursprungligen användes fotografisk registrering av spektrum, men i dag är detta inte aktuellt när det gäller rutinanalyser. I stället används fotomultiplikatorer (PMT), som det finns ett antal olika typer av, med varierande energikänslighet. Fotodioder kan i princip också användas, men dessa har lägre känslighet i det låga våglängdsområdet.
Man har även börjat använda fotodiod-array med stort antal dioder (pixels) sammansatta till en ”elektronisk film”. Andra benämningar på denna typ av detektorer är CCD (Charge-Coupled-Device) och CID (Charge-Injection-Device). Med sådana detektorer finns möjlighet att göra såväl simultan bakgrundsmätning som profilering.
De små detektorelementen (20–30 μm) kräver en tvådimensionell fokusering av spektrum. Detta kan erhållas med hjälp av en så kallad Echelle spektrometer. Känsligheten för solid-state-detektorer är dock fortfarande sämre än för fotomultiplikatorer.
Fotomultiplikatorn alstrar en ström som är proportionell mot ljusintensiteten hos spektrallinjen. Genom att låta den erhållna strömmen ladda en kondensator, kommer laddningen över den att motsvara ett ”medelvärde” på strömmen under mättiden. Detta medför att spänningen över kondensatorn är proportionell mot ljusintensiteten och även mot elementkoncentrationen. Nyare spektrometrar har elektronik med direkt integrering utan kondensatorer.
Kalibrering och standardisering
Vid grundkalibrering av en optisk spektrometer för prov i fast form kan man inte använda rena kemikalier, utan man är hänvisad till referensmaterial (RM) med kända elementkoncentrationer, eventuellt kompletterat med egna prover, som analyserats med andra metoder.
Vid kalibreringen erhålls för varje element och referensprov ett intensitetsvärde I, som tillsammans med provets uppgivna koncentration C, matas in i spektrometerdatorn. En kalibrering för stålanalyser kan kräva hundratals referensprov, och det blir således ganska stora datamängder att hantera. Ett regressionsprogram beräknar kurvfunktionen för respektive element. Det är sällan funktionen är linjär, utan i regel handlar det om andra- eller tredjegrads polynom. Ett tredjegrads polynom får följande utseende:
C = a0+a1 · I+a2 · I2+a3 · I3
där an är koeffcienter erhållna vid regressionen. För att öka stabiliteten använder man inte ett elements absolutintensitet utan kvoten mellan intensiteten för elementet och intensiteten för en stabil linje av baselementet, i detta fallet järn, det vill säga I = I(el)/I(Fe). Eventuella interelementeffekter beror vanligen på linjestörning och/eller matrisinterferens, vilka man måste korrigera för. Korrektionstermerna beräknas i regressionen.
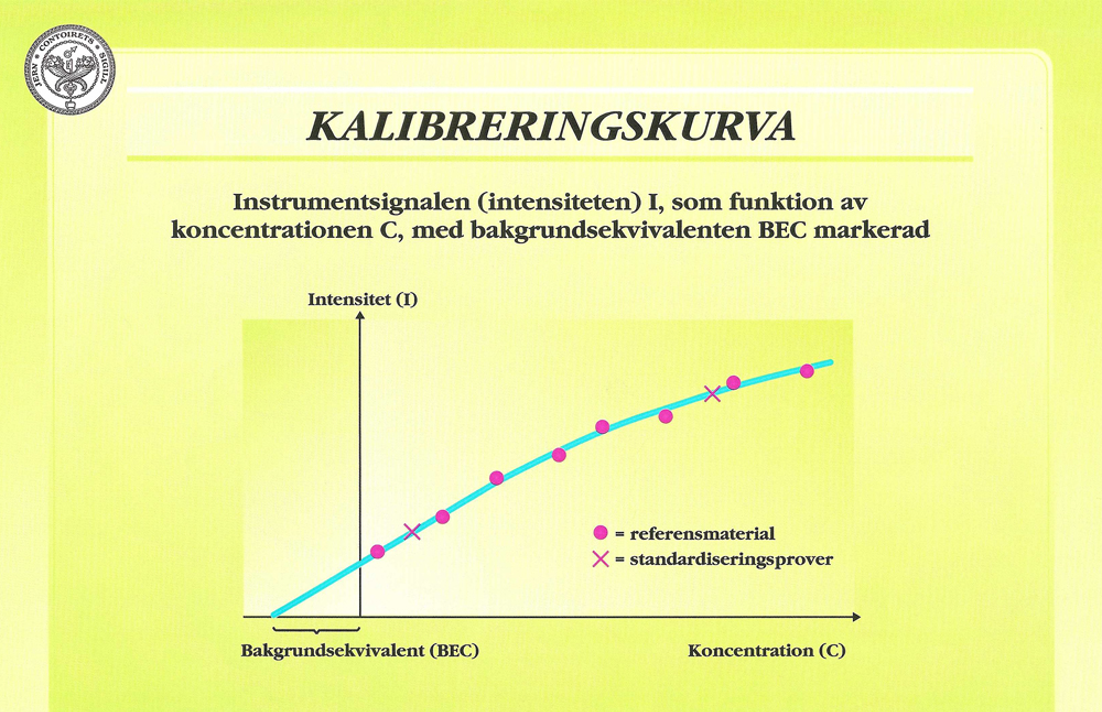
Beroende på bland annat nedsmutsning av gniststativ och lins samt eventuella variationer i elektriska kretsar, kan kalibreringen ändras och måste därför kontrolleras med vissa intervall, till exempel efter ett antal prover eller med vissa tidsintervall. Detta sker genom att analysera standardiseringsprover (engelska: setting up samples), vanligen ett lågprov och ett högprov. I Figur 27 visas en typisk analyskurva, där kalibreringsunderlaget (RM:s och CRM:s) markerats med punkter och standardiseringsproven med kryss.

Tillåtna intervall för standardiseringsproven läggs in i spektrometerdatorn, som larmar om resultatet av standardiseringen går utanför gränserna.
Analysförfarande
Spektrometern kan förses med ett antal analysprogram med olika gnistparametrar, elementkanaler och koncentrationsområden. I och med att spektrometern är kalibrerad och inställd, är det mycket enkelt att använda den.
Analysgången är följande:
- renborstning av elektrod
- fixering av provet i gniststativet
- val av analysprogram
- initieringavprovidentitet
- start gnistning nr 1
- flyttaprovet
- start gnistning nr 2
- kontroll av utskrift
- eventuell ytterligare gnistning (till exempel vid dåligt prov) eller komplettering av element bestämt på annat sätt
- rapportering
Enkelheten i förfarandet inbjuder till automatisering, vilket också sker i allt större utsträckning, se vidare kapitel 7.
Bakgrundsekvivalent (BEC) och detekterbarhetsgräns (DL)
Beroende på att en viss mängd ströljus alltid kommer in i spektrometern, ger detta en bakgrund under själva spektrumet. Bakgrunden bestämmer i stor utsträckning med vilken precision man kan bestämma låga halter av ett element och har således stor betydelse för detekterbarhetsgränsen. Ett vanligt sätt att uttrycka detta är att ange bakgrundsekvivalenten BEC (Background Equivalent Concentration), vilken definieras som den koncentration av elementet som ensamt skulle ge samma ljusintensitet som bakgrunden.
På motsvarande sätt definieras detekterbarhetsgränsen DL (Detection Limit) som den koncentration som är lika med tre gånger standardavvikelsen för bakgrunden. Som framgår av Figur 27 erhålls värdet på BEC direkt genom att extrapolera analyskurvan. Instrumenttillverkarna anger ofta DL för olika element i sina broschyrer.
Sambandet mellan BEC och DL brukar uttryckas i formeln DL = 3 · RSD · BEC, där RSD motsvarar relativa standardavvikesen för bakgrundssignalen. I praktiken har det visat sig att BEC vanligen är 20-30 gånger större än DL. BEC och DL är mycket användbara för att kontrollera en spektrometers status och för att jämföra olika instrument. Man skall dock inte sätta likhetstecken mellan DL och den lägsta koncentration av ett element som man kan rapportera. Denna halt kallas undre bestämbarhetsgräns och bör sättas 3-5 ggr DL.
2.3.2 Optisk emissionsspektrometri med glimurladdningslampa (GDOES)
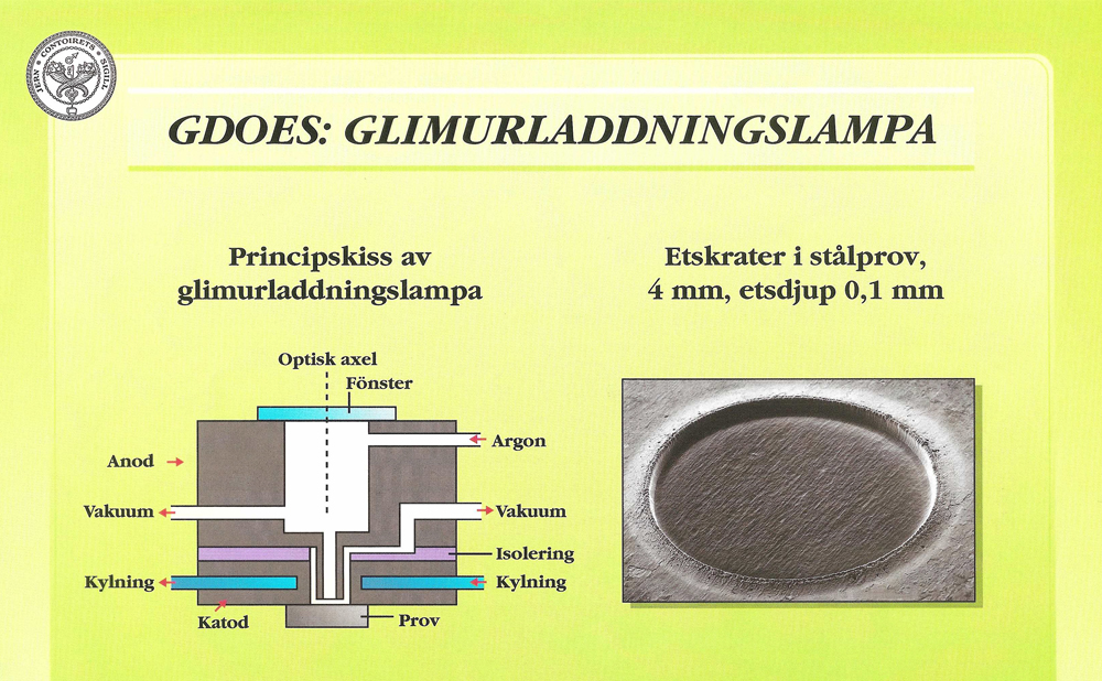
Som alternativ till gniststativ har under senare år glimurladdningslampa (GDL, Glow Discharge Lamp) börjat användas allt mer. Den första glimurladdningslampan konstruerades av W. Grimm redan på 1960-talet. Det är en ljuskälla baserad på principen lågtrycksurladdning i ädelgasatmosfär. Figur 28:a visar konstruktion och princip av en GDL.
Provet, som måste vara plant och elektriskt ledande, får utgöra katod i lampan. Anoden är utformad som ett rör med 7 – 8 mm innerdiameter, och dess mynning befinner sig omedelbart (0,2 mm) framför provet. Urladdningsrummet evakueras med en kapselpump och genomspolas därefter med argon vid ett tryck av cirka 130 Pa.

Då en spänning av storleksordningen 500 – 1000 V anbringas, uppstår en elektrisk urladdning. Ett område på den katodiska provytan, som motsvarar anodöppningen, bombarderas av positivt laddade joner från plasmat. Provytan jonetsas av urladdningen (engelska: cathodic sputtering). En del av det bortetsade provmaterialet exciteras i glimskiktet och ger upphov till optisk emission av de ingående elementens karakteristiska våglängder.
Då jonetsningen är jämn över brännfläcken, åstadkoms en krater med i stort sett plan botten. Figur 28:b visar ett foto av en etskrater i ett stålprov. Diametern är 4 mm och etsdjupet 0,1 mm.
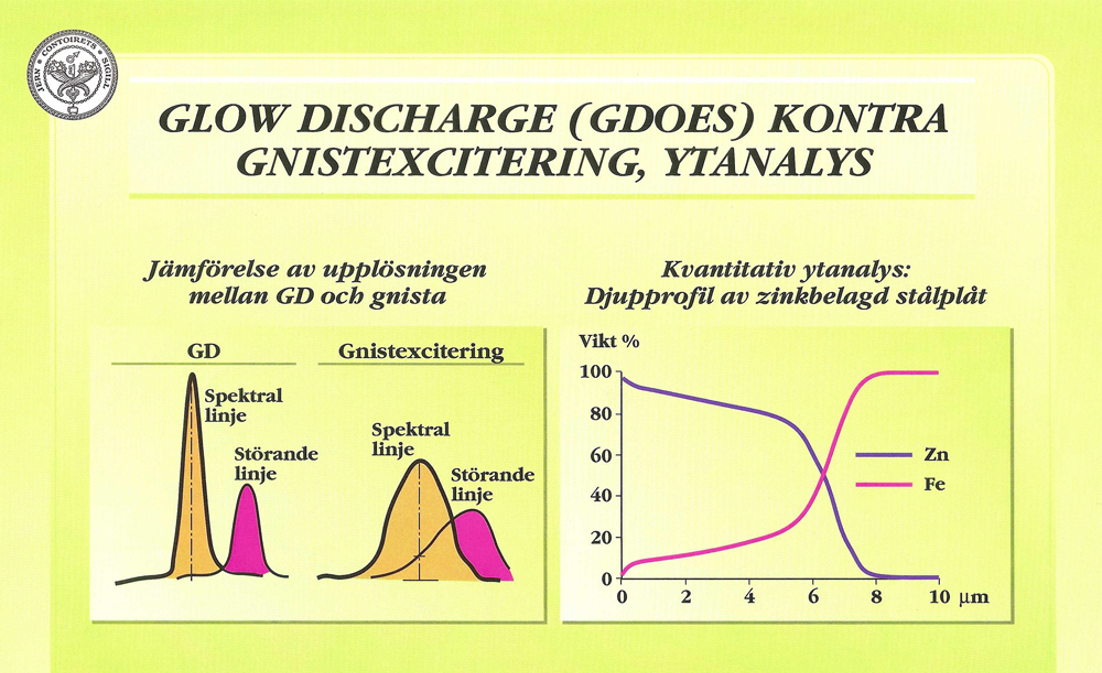
En undersökning har utförts vid Institutet för Metallforskning (IM) i Stockholm för att jämföra gniststativ med GDL. Undersökningen visar flera fördelar när det gäller GDL, bland annat följande:
- mindre känslig för matriseffekter och linjeinterferenser, se Figur 29:a
- linjära analyskurvor upp till höga legeringshalter
- lägre detekterbarhetsgränser för många vanliga element
Nackdelen med GDL i jämförelse med gniststativ är något längre analystid och omständligare hantering, bland annat på grund av noggrann provberedning.

Ytanalys /djupprofilanalys med GDL
Då urladdningsrummet i glimurladdningslampan hela tiden evakueras, befinner sig i varje ögonblick i plasmat provmaterial härrörande från endast ett fåtal atomlager.
Dessa egenskaper gör glimurladdningslampan lämplig för ytkemisk analys, eftersom man kan följa intensitetsförändringar från olika element i plasmat allteftersom jonetsningen av provytan fortgår. I jämförelse med andra ytanalystekniker, såsom Auger och SIMS, är GDL enklare och snabbare att tillämpa.
Från början användes ytanalys med GDL mest i kvalitativt hänseende. Efter grundläggande undersökningar vid IM, har man där utvecklat system även för kvantitativ ytanalys (djupprofilanalys), vilket har stor betydelse för kvalitetskontroll vid ytbeläggning av stål och metaller. Andra tillämpningar är studium av gränsskikt och utarmade zoner. Med djupprofilanalys kan skikt mellan några nm och cirka 150 μm studeras. Denna metod har vunnit internationell uppmärk- samhet. Figur 29:b visar en djupprofil av zinkbelagd stålplåt.
2.3.3 Röntgenfluorescensspektrometri, XRF
Medan OES med gnistexcitering är begränsad till analys av metalliska prover, har XRF fördelen att kunna användas för analys av praktiskt taget alla typer av material, såväl metalliska som oxidiska. Proven kan vara kompakta eller pulverformiga, och med särskilda provhållare kan även vätskor analyseras.
En annan fördel med XRF är att metoden i princip är oförstörande, vilket bland annat är positivt med tanke på minskad förbrukning av kalibrerings- och standardiseringsprover. En modern röntgenspektrometer har hög stabilitet, vilket bidrar till att precisionen vid XRF-analys är bättre än andra instrumentella analysmetoder. Röntgenmetodens begränsningar är främst dålig detekterbarhet vid bestämning av lätta element (Z < 12), även om prestanda i detta avseende har förbättrats under senare år.
De första röntgenspektrometrarna kom i praktiskt bruk i slutet av 1950-talet. I dag är XRF-tekniken allmänt tillämpad vid stålverkslaboratorier och vid andra laboratorier som arbetar med oorganiska analyser.
Röntgenstrålningens uppkomst
Elektronerna i en atom befinner sig i bestämda energitillstånd, så kallade elektronskal, vilka från atomkärnan räknat benämns K, L, M, N – – – , se Figur 30:a. Vi har tidigare konstaterat att synligt och ultraviolett ljus alstras genom elektronövergångar i atomernas yttre skal. Ju längre ut från kärnan elektronen befinner sig, desto lättare är det att påverka den (mindre energi behövs). När det gäller röntgenstrålning är det elektronövergångar i skalen närmast atomkärnan som är upphovet, och för detta krävs hög energi.
När ett material bombarderas med elektroner av hög hastighet eller bestrålas med energirika fotoner, finns en viss sannolikhet att någon av de elektroner som befinner sig närmast atomernas kärnor skjuts bort och efterlämnar ett tomrum.

En elektron från något av de yttre skalen intar då den tomma platsen. I den yttre banan hade elektronen emellertid högre energi än i den nya inre banan, och energiskillnaden kommer att sändas i form av elektromagnetisk strålning. Om våglängden hos denna strålning är mellan 0,01 och 10 nm, kallar vi den röntgenstrålning.
Varje möjligt elektronsprång mellan de olika energinivåerna ger upphov till strålning av bestämd energi, vilken är karakteristisk för atomen i fråga. Denna energimängd är proportionell mot strål- ningens frekvens υ, och alltså omvänt proportionell mot dess våglängd, λ. Den alstrade röntgen- strålningen benämns efter mellan vilka skal elektronövergångarna sker, till exempel Kα, Kβ, Lα och så vidare, Figur 30:a åskådliggör detta.
Röntgenröret
En skiss av ett röntgenrör visas i Figur 30:b. Principen är i korthet att katoden, som är utformad som en glödtråd upphettas genom en elektrisk ström. Genom att lägga en hög spänning mellan katod och anod, accelereras elektroner mot anoden. Vid elektronernas uppbromsning i anodmaterialet genereras röntgenstrålning, vilken visar sig dels som en kontinuerlig strålning och dels som en karakteristisk strålning, det senare under förutsättning att spänningen är tillräckligt hög. Den karakteristiska strålningens våglängd är beroende av anodmaterialet. För att röntgenröret skall fungera måste det evakueras till högvakuum.
Det vanligaste anodmaterialet i dagens röntgenrör är rodium, men även krom, molybden och volfram förekommer. Effekten brukar vara 3 kW med en maxspänning av 60 kV och en strömstyrka upp till 125 mA. Den höga effekten gör att röret måste kylas. Figur 30:c visar ett modernt röntgenrör med ändfönster, vilken konstruktion gör att avståndet mellan anod och prov kan minimeras och därigenom göra exciteringen effektivare. Röntgenrörets fönster brukar vara tillverkat av beryllium, 75 – 150 μm tjockt.
Absorption och excitering av röntgenstrålning
Detta avsnitt skulle behöva en mer ingående förklaring, vilket utrymmet tyvärr inte medger. I stället får vi nöja oss med att konstatera följande fakta. Varje grundämne absorberar röntgenstrålning på ett sätt, som är karakteristiskt för ämnet i fråga och som är beroende av röntgenstrålningens våglängd.
Absorptionen i ett ämne brukar uttryckas genom ämnets massabsorptionskoefficient. Figur 31:a visar hur massabsorptionskoefficienten för nickel varierar med röntgenstrålningens våglängd. Absorptionsförmågan stiger till en början med våglängden, men sjunker abrupt vid cirka 0,15 nm, för att sedan återigen gradvis öka.
Vid högre våglängder inträffar motsvarande fenomen för L- och M-strålning. Man talar om respektive K-, L- och M-absorptionskanter.

Om vi nu vill bestämma nickel genom att excitera dess karakteristiska strålning (Kα) med strålning från ett röntgenrör, fordras att den infallande strålningen har högre energi än den alstrade, det vill säga endast den del av spektrum från röntgenröret som har kortare våglängd än nickels absorptionskant (0,148 nm) bidrar till excitationen. Av det sagda framgår att man genom val av rönt- genrör med lämpligt anodmaterial, kan optimera exciteringsbetingelserna. Högst intensitet erhålls om en karakteristisk linje i rörspektrat ligger strax till vänster om absorptionskanten på det element som skall bestämmas.
Genom att öka spänningen på röntgenröret, ökar intensiteten inte enbart på grund av ökad effekt, utan också genom att rörets kontinuerliga spektrum förskjuts mot kortare våglängder. En ökning av strömstyrkan påverkar däremot intensiteten linjärt.
Upplösning av röntgenspektra
För att kunna studera intensitets- och energifördelningen hos röntgenstrålning, måste man kunna dela upp den i dess spektrala komponenter i likhet med vad fallet är när det gäller OES. På grund av den korta våglängden hos röntgenstrålningen, är det inte möjligt att använda gitter eller prisma som dispersivt medium. I stället används enkristaller med väldefinierade atomplan parallella med ytan. När polykromatisk röntgenstrålning träffar kristallen reflekteras endast den våglängd som uppfyller villkoret i Braggs ekvation
nλ = 2d · sinθ
där n = strålningens ordningstal, λ = våglängden, d = avståndet mellan kristallens atomplan och θ = vinkeln mot kristallplanet.
Den reflekterade strålningen är således monokromatisk. Avståndet mellan atomplanen (d-värdet) samt vinkeln hos den infallande strålningen är avgörande för hur monokromatiseringen sker. Detta framgår av Figur 31:b. Vanliga kristaller är LiF (litiumfluorid), Ge (germanium), PE (penta- erytritol), ADP (ammoniumdivätefostat) och syntetiska så kallade multilayerkristaller, alla med olika d-värden.
Röntgenspektrometrar
I en våglängdsdispersiv röntgenspektrometer ingår följande huvudkomponenter:
- röntgenrör
- filter
- provväxlare med provkassetter
- kollimatorer eller spalter
- kristaller
- detektorer
Även när det gäller röntgenspektrometrar används såväl sekvensiella som parallellmätande instrument. Figur 32:a-d visar schematiskt strålgång och utförande i sådana spektrometrar. Gemensamt för båda typerna är röntgenrör, filter och provväxlare med provkassetter. Filter mellan röntgenrör och prov måste användas, om röret utsänder karakteristisk strålning, som starkt stör ett element som skall bestämmas. Materialet i filtret väljs så att dess absorptionskant ligger strax över den störande linjens våglängd.

I den sekventiella spektrometern vrids den platta kristallen så att dess yta och atomplan kommer i reflektionsläge, det vill säga uppfyller villkoret i Braggs ekvation. Observera att detektorns rörelse blir dubbelt så stor som kristallens. Kollimatorernas funktion är att rikta strålningen mot kristall respektive detektor. Kombinationen kollimatorer, kristall och detektor i en sekvensiell spektrometer kallas goniometer.
I den parallellmätande spektrometern används spalter i stället för kollimatorer för att avgränsa strålningen. Dessutom är lägena för spalter, kristall och detektor fixerade för varje element. Varje sådan uppsättning benämns monokromator. För att öka intensiteten används böjda kristaller, som fokuserar den monokromatiska strålningen mot detektorn. En parallellmätande spektrometer har i regel plats för upp till 24 monokromatorer. En del tillverkare har spektrometrar med såväl sekvensiell som simultan funktion. För att kunna bestämma lätta element med långa våglängder, måste spektrometern hållas under vakuum. Figurerna i Figur 33:a-b visar två moderna röntgenfluorescensspektrometrar av parallellmätande och sekvensiell typ.

Detektorer
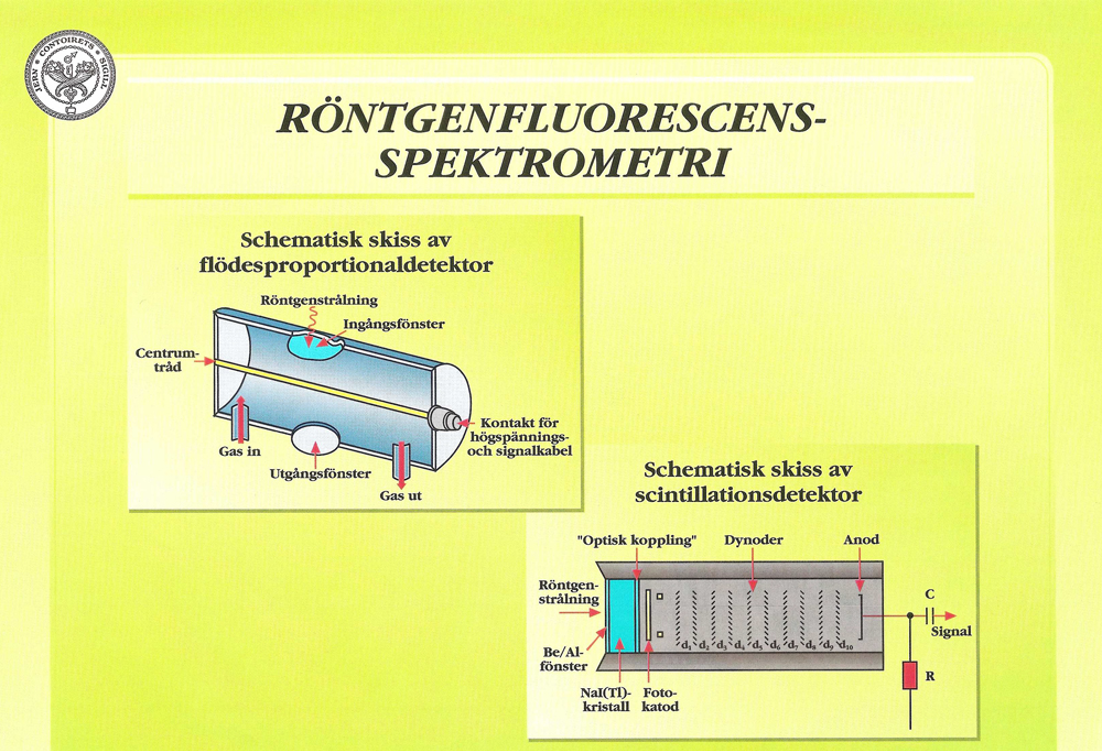
För att registrera såväl långa som korta våglängder används två typer av detektorer; flödesproportionaldetektorn (flödesdetektorn) för långvågig strålning och scintillationsdetektorn för kortvågig. Figur 34:a visar schematiskt en flödesdetektor. Den består av en kammare genom vilken en metalltråd är spänd. Detektorn har fönster av tunn aluminiumbelagd polypropylen. På tråden läggs en spänning av cirka 1000 V. En gasblandning av argon/metan (90/10 %) får långsamt strömma genom detektorn.

Då ett röntgenkvantum träffar detektorn sker en jonisering av gasmolekylerna och en puls genereras via centrumtråden till signalkabeln och en förstärkare. Amplituden hos en urladdning är proportionell mot energin hos det infallande röntgenkvantat, och därigenom blir pulshöjden karakteristisk för den infallande strålningens energi. Ofta kopplas en flödesdetektor i serie med en scintillationsdetektor; därav två fönster på flödesdetektorn i Figur 34:a.
Scintillationsdetektorn bygger på en annan princip, vilket framgår av Figur 34:b. Där ingår först och främst en talliumdopad NaJ-kristall, vilken vid röntgenbestrålning exciteras och avger ljuskvanta av cirka 400 nm våglängd. De genererade ljusblixtarna representerar var och en ett röntgen- kvantum, som registreras av en fotomultiplikator uppbyggd av en serie så kallade dynoder. Pulserna från detektorn förstärks och matas in i spektrometerns mätelektronik på vanligt sätt. Även scintillationsdetektorn avger elektriska pulser, som är proportionella mot den infallande röntgenstrålningens energi.
Förutom redan beskrivna detektorer används även slutna proportionalitetsdetektorer. Skillnaden mellan dessa och en flödesdetektor är, förutom att de är slutna, att de kan vara fyllda med andra ädelgaser än argon, till exempel neon, krypton eller xenon. Dessutom brukar detektorfönstret vara av beryllium. I parallellmätande spektrometrar är dessa detektorer vanliga.
Pulshöjdsdiskriminering
Den strålning som kommer in i detektorn vid en viss vinkelinställning kan ha annat ursprung än det avsedda. Av Braggs ekvation framgår att en våglängd i första ordningen av exempelvis 1 nm, hamnar på samma vinkel som våglängden 0,5 nm i andra ordningen. Även andra linjer som ligger nära den som skall mätas kan störa genom att upplösningen inte räcker. Men tack vare olika energi hos strålningen, och därigenom olika amplitud hos pulserna från detektorn, kan man på elektronisk väg välja vilka pulser som skall registreras. Man ställer in en ”tröskel” och ett ”fönster” på pulshöjdsdiskriminatorn och då räknas endast de pulser vilkas amplitud ligger inom fönstret.
Linjeinterferenser och matriseffekter
Linjeinterferenser, där det endast föreligger små energiskillnader, går som vi sett inte att eliminera genom pulshöjdsdiskriminering. I stället får man tillgripa matematisk korrektion i likhet med OES. Med få undantag är sådana störningar sällan något problem att komma tillrätta med när det gäller XRF.
Störningar på grund av matriseffekter är däremot mer komplicerade, då man nästan alltid måste ta hänsyn till skillnader mellan olika provers absorption av såväl den primära strålningen från röntgenröret som den sekundära strålningen från själva provet.
Dessutom måste hänsyn tas till sekundär excitering, vilket innebär att ett eller flera elements karakteristiska strålning exciterar det element, vars koncentration skall bestämmas, och därigenom ökar fluorescensstrålningen från detta element.
Att beräkna matriskorrektioner genom regressionsanalys kräver vanligen ett mycket stort antal referensprover med tillräckligt stora koncentrationsintervall. Därför är det vanligt att beräkna teoretiska korrektionsfaktorer baserade på en genomsnittlig sammansättning. Dessa korrektionsfaktorer gäller för ett begränsat område.
Kalibrering och standardisering
Principen för kalibrering och standardisering överensstämmer i stort med den som gäller OES, kapitel 3.3.1. En röntgenspektrometer är dock ett mer komplicerat instrument än en optisk spektrometer och kräver därför mer inställning och kontroll, speciellt om det är en sekvensiell spektrometer. Exempel på sådana inställningar är 2θ-vinkeln, detektorspänning, pulshöjdsfördelning, effekt på röntgenröret samt mättider. I övrigt är kalibreringsförfarandet detsamma som vid OES med mätning av ett stort antal kalibreringsprov och minst två standardiseringsprov per element (hög- och lågprov).
Kalibrering enligt fundamentalmodell
Detta är en annorlunda kalibreringsteknik som utvecklats på senare år. I korthet går fundamentalmodellen ut på att man endast en gång behöver utföra grundkalibrering genom att bestämma spektrometerns känslighet för varje aktuellt element genom att mäta på rena substanser.
Alla element som kan tänkas ingå i ett prov måste mätas för att modellen skall fungera. Dessutom bestäms en kontinuerlig bakgrundskurva genom att mäta på en organisk substans, till exempel teflon, vid olika våglängder. All övrig information om vad som händer med röntgenstrålningen i provet finns i programmet, såsom massabsorptionskoefficienter, korrektioner för interelement- och matriseffekter et cetera.
Vid analys samlas råintensiteter i ett hundratal kanaler och programmets uppgift är att från dessa beräkna nettointensiteter för varje kanal och att därefter beräkna halterna med hjälp av känslighetsfaktorerna.
Med fundamentalmodellen kan praktiskt taget alla typer av material analyseras. Bäst resultat erhålls om provet täcker hela den exponerade ytan samt är ”oändligt” tjockt med avseende på röntgenstrålningen. Det går även att få acceptabelt resultat (halvkvantitativ analys) på prover med begränsad yta och tjocklek, särskilt om man matar in information om provet i programmet före analys.
Utvärderingsprogram, som bygger på fundamentalmodellen, levereras av spektrometertillverkarna bland annat under namn som UniQuant och SemiQuant.
Detekterbarhetsgräns (DL)
I kapitel 3.3.1 definieras BEC och DL. Samma definitioner gäller vid röntgenanalys, men här brukar man också använda följande statistiska formel: 
där S är känsligheten i CPS/% (pulser per sekund och procent) och Rb bakgrunden i cps samt T mättiden i sekunder. Vanligen används 100 sekunders mättid. Denna formel tar endast hänsyn till den statistiska spridningen. För att kontrollera konditionen hos spektrometern och vid jämförelse med andra instrument är den dock användbar.
Provpreparering
Provberedning av råjärn och stål för spektrometrisk analys har berörts i kapitel 2.8.2 och 2.9.3. Här kan endast understrykas att när det gäller provberedning av metalliska prov för XRF-spektrometrisk analys, är det speciellt viktigt att åstadkomma en fin och reproducerbar ytstruktur.
När det gäller oxidiska material har vi förut behandlat provberedning fram till så kallat analysfint prov, det vill säga prov nedmalt till < 70 μm. Det är fullt möjligt att anlysera ett sådant prov direkt genom att hälla provpulvret i en provkopp med mylarfilm, som är genomsläpplig för röntgenstrålning. Förutsättningen är att bestrålningen sker underifrån vilket brukar vara fallet med sekventiella instrument.
Ett vanligare och bättre sätt är att pressa provet till en brikett med eller utan bindemedel. Man får då ett kompakt prov, där variationer i kornstorlek inte påverkar resultatet lika mycket. Man kan dock inte bortse från sådana störningar, så det gäller även här att kunna reproducera provberedningen.
Bästa sättet att få ett homogent prov är att smälta (uppsluta) det i litium- eller natriumtetraborat och gjuta provet till en platta. Man får då ett glas, som kan betraktas som en fast lösning. Detta är en metod som tillämpas alltmer. En stor fördel med den är att man kan utgå från rena kemikalier vid kalibrering. En vanlig utspädning är 5 à 10 gånger.
2.3.4 Bestämning av kol och svavel i stål med förbränningsanalys
Även om man med god noggrannhet kan bestämma kol och svavel med OES och svavel med XRF, är det ofta nödvändigt att komplettera en sådan bestämning med en förbränningsanalys på ett separat prov. Detta gäller särskilt vid extremt låga halter (< 100 ppm).
En annan anledning kan vara att man önskar uppnå bättre provrepresentativitet, vilket sker vid förbränningsanalys, tack vare att man använder större provmängd per bestämning, vanligen 1 g. Detta skall jämföras med den analyserade massan av endast något mg vid en spektrometrisk analys.
Analysatorer finns dels i kombinerat utförande för kol/svavel och dels som separata instrument för dessa element. En C/S-analysator består av en ugnsenhet och en analysatorenhet var för sig eller sammanbyggd i en enhet. Analysprincipen är i korthet att provet förbränns i syrgas varvid kol och svavel oxideras till CO2 och SO2, som mäts i IR-detektorer.
Ugn och förbränning
Vid analys av stål och metaller används i dag nästan alltid högfrekvensugnar. Vid analys av icke metalliska produkter används däremot motståndsugnar. Effekten för en HF-ugn är vanligen cirka 2 kW och frekvensen 18 MHz. Provet, i form av spån eller hel bit, förbränns i en keramisk degel i en ström av ren syrgas. Degeln skyddas av ett kvartsrör som omges av HF-slingan.
För att underlätta förbränningen tillsätts vissa acceleratorer såsom koppar, tenn, järn eller volfram i granul- eller spånform. Vid förbränningen uppnås en temperatur av cirka 2900°C. Förbrän- ningsgaserna innehåller CO2, CO, SO2 och H2O samt stoft i form av metalloxider. Gaserna renas från stoft i en porös fälla och från fukt i en fälla med magnesiumperklorat.
Analysatorn
Via en flödeskontroll leds de renade förbränningsgaserna först till en IR-detektor där SO2-halten mäts. Därefter leds gaserna genom en katalysatorugn, som oxiderar CO till CO2 och SO2 till SO3, vilken senare adsorberas i en cellulosafälla. CO2 leds vidare till en andra IR-detektor, som mäter absorbansen.
I analysatorer för ultralåga kolhalter finns en tredje IR-detektor med längre strålgång. Analysområdet för ett instrument med två kolintervall uppges till 0,6 ppm – 0,5 % respektive 6 ppm – 6 % samt för svavel 0,6 ppm – 0,35 %. Precisionen uppges för båda elementen till 0,3 ppm eller 0,5 %, vilketdera som är högst.
Det förekommer även kolanalysatorer med TC-detektor i stället för IR. Denna typ av analysator får dock längre analystid, cirka 2 min mot cirka 40 sek för instrument med IR-detektor. Figur 35:a visar ett flödesschema på en modern kol/svavelanalysator och Figur 35:b instrumentutförande.

Kalibrering – analys
Kalibreringen kan utföras endera med rena gaser eller med referensmaterial. Då lineariteten är god kan så kallad single point kalibrering tillämpas. Blankvärdet måste dock alltid kontrolleras. Tack vare hög grad av automatik är utförandet av en analys mycket enkel. Efter invägning är det i princip endast att trycka på startknappen. Analystiden är 40-50 sekunder. Det är emellertid synnerligen viktigt att rengöra förbränningsrör och byta fällor regelbundet.
2.3.5 Bestämning av kväve och syre med smältextraktion
Kväve är numera möjligt att bestämma med OES, men känsligheten är i underkant för låga halter. Därför hör kvävebestämning med smältextraktion till de vanligaste analyserna vid ett stålverkslaboratorium. Syrebestämningar är däremot inte lika frekventa, ofta utförs dessa mer kampanjvis.
Liksom i fallet kol/svavel är det vanligt att man kombinerar kväve- och syrebestämning i samma instrument. Detta är dock mera tveksamt, eftersom syre kräver annan provberedning än kväve (mer om detta senare). Separata analysatorer för kväve och syre finns även. En kväve/syre analysator brukar också bestå av två enheter: ugn- och analysatorenhet. Analysprincipen går ut på att provet smälts i en grafitdegel varvid kväve extraheras som N2 och syre reagerar med kol till CO, som oxideras till CO2. De bildade gaserna leds med helium till varsin detektor, IR-detektor för CO2 och TC-detektor för N2. Vissa instrumenttyper mäter CO direkt med IR-detektor.
Ugnen
Ugnen består av två kraftiga vattenkylda elektroder. På den undre elektroden placeras en grafitdegel, som sedan trycks mot den övre elektroden. Degeln upphettas genom att en hög ström leds genom den. Strömstyrkan kan variera mellan 600 och 1300 A, varvid temperaturer upp till 3000ºC kan uppnås. Degeln avgasas först vid högre temperatur, varefter provet slussas in och temperaturen höjs igen så att provet smälter. Därvid extraheras kväve och syre från provet. Tack vare en kemisk reaktion med grafitdegeln, behöver syre inte lika hög temperatur som kväve för att extraheras kvantitativt.
Analysatorn
Från ugnen leds de extraherade gaskomponenterna med helium till analysatorn, där de passerar följande enheter: katalysatorugn, där CO oxideras till CO2, flödeskontroll, IR-detektor för bestämning av CO2-halten (det vill säga syrehalten), fällor för absorption av CO2 och eventuell fukt samt slutligen en TC-detektor för bestämning av kvävehalten. Alla funktioner styrs och kontrolleras av en mikroprocessor.
Typiskt analysområde vid 1 g provvikt är för syre 0,1 – 1000 ppm och för kväve 0,1 – 500 ppm. Precisionen uppges för syre till 0,5 ppm eller 1 % och för kväve till 1 ppm eller 1 %, vilketdera värde som är högst. Figur 35:c visar flödesschema på en kväve/syreanalysator och Figur 35:d en figur på en sådan.
Kalibrering – Analys
Analysatorn kan kalibreras med rena gaser eller med referensmaterial. Bäst är att använda båda sätten, då man vid överensstämmelse mellan kaliberingsmetoderna får bevis för att instrumentet är i god kondition. Själva analysen är lika enkel som för kol/svavel, det vill säga tryck på knappen och invänta analysresultat. Analystiden är något längre än för kol/svavel på grund av degelavgasning. Viktigt för funktionen är även här rengöring av ugn och byte av fällor med regelbundna intervall.
Att tänka på före syrebestämning
Till skillnad från kväve är syre ojämnt fördelat i stålet. I smält stål föreligger syre dels löst och dels bundet som oxider. Om avsikten är att bestämma totala syrehalten i smält stål, måste därför provet tätas väl med aluminium. För att undvika makroslagger är det en fördel att ta bulkprov, där stålet stelnar långsamt så att de största slaggerna hinner flyta upp till ytan. Av tidsskäl används i dag dock mest glaspipett för syreprovtagning av smält stål. Behovet av provtagning av smält stål för syrebestämning har dock minskat tack vare användning av syresonder för löst syre.
I det färdiga materialet är det av stort intresse att ha kontroll på syrehalten, eftersom denna är ett mått på stålets renhet. Även om inte varje charge kontrolleras är det viktigt att göra det vid tillverkning av nya stålsorter och vid förändring av processerna. Provtagningen sker genom sågning eller kapning, se vidare avsnitt 2.9.2. Spån kan ej användas. Vid provberedningen är det viktigt att avlägsna alla ytoxider. Detta kan ske genom filning eller annan mekanisk bearbetning. Provet får dock inte bli varmt. Före analys avfettas och torkas provet.
2.3.6 Bestämning av väte i flytande stål
Det är viktigt att bestämma vätehalten i stål på grund av risk för sprickor (flakes) i det färdiga materialet om vätehalten är för hög. Detta gäller i första hand stål med låga legeringshalter. Höglegerade stålsorter, till exempel rostfritt, är inte utsatta på samma sätt tack vare hög löslighet av väte även i fast tillstånd.
Väte i flytande stål kan bestämmas med olika metoder. Följande tre metoder är de vanligaste:
- Uttag av prov med vakuumpipett av glas, kylning och frysning av provet. Bestämning av vätehalten genom varm- eller smältextraktion.
- Provtagning med så kallad engångs vakuumprovtagare av stål. Med denna provtagare erhålls dels ”diffunderbart” väte och dels restväte som kan bestämmas med varm- eller smältextraktion.
- Direktmätning med sond. Mätningen baseras på jämvikt av partialtryck av väte i stålsmältan och en inert gas som spolas genom smältan.
Metod 1: Vakuumpipett av glas
Denna metod är den klassiska metoden för vätebestämning i stål. Efter provtagning kyls provet i vatten och fryses i kolsyreis eller flytande kväve/alkoholblandning. Tidigare togs provet med skopa och göts i kopparkokill för snabb kylning, men detta förfarande har i dag ersatts med vakuumpipett som är enklare att hantera. Pipetten kan fästas i en enkel hållare eller monteras i en sond. Viss risk finns dock för väteförluster i samband med provtagning och provberedning.
Det är viktigt med en noggrann provberedning: provet måste ha en ren yta, eventuella oxider avlägsnas med filning eller slipning.
Bestämningen av väte kan göras med varmextraktion i en analysator med ugn av induktions- eller motståndstyp. Provvikten kan variera mellan 5 och 10 g. En extraktionstemperatur av 900 – 1000ºC räcker för de flesta ståltyper. Vid denna temperatur brukar en analystid av cirka 10 minuter vara tillräcklig för att extrahera ut allt väte.
Ett alternativ till varmextraktion är smältextraktion vid en temperatur av 1900 – 2800ºC. Detta är en snabbare metod, analystid cirka 3 minuter. För vissa specialstålsorter samt för metaller såsom titan och tantal, där varmextraktion är otillräcklig, är smältextraktion att föredra. Detektorn i väteanalysatorer är av TC-typ (termisk konduktivitet). Vid smältextraktion, där även kväve frigörs, sker separation med en molekylsikt.
Metod 2: Engångs vakuumprovtagare av stål (ESK)
Figur 36:a visar principen för denna provtagare, som brukar kallas ESK av tyska ”Einweg Saug Kokille”. Fördelen med denna provtagare är att man undviker förluster vid provtagningen. Provtagaren består av en evakuerad stålhylsa, som innehåller en rörformad kokill. Vid neddoppning i stålsmältan tränger stål in i kokillen. Under stelnandet diffunderar väte från provet till det omgivande hålrummet och stannar där. Stålhylsan kyls i vatten direkt efter provtagningen.

Det diffunderade vätet i form av H2 mäts i en analysator, Figur 36:b, genom punktering av stål- hylsan och spolning med luft för överföring av vätet till en TC-detektor. Analysatorn kalibreras med ren vätgas eller helium. Bestämningen tar endast 1 à 2 minuter.
I det stelnade provet finns en viss mängd restväte, som kan bestämmas på konventionellt sätt med varm- eller smältextraktion. Restvätehalten är beroende av stålsorten, ju mer legerat materialet är desto högre blir restvätehalten. Vanligen är restvätehalten 10 – 30 % av totala vätehalten. Tack vare god reproducerbarhet kan man oftast införa en korrektionsfaktor för restvätet, vilket gör metoden mycket enkel och attraktiv att använda.
Metod 3: Direktmätning med sond (Hydris)
Direktmätning av vätehalten i en stålsmälta har länge varit ett önskemål och försök därtill har utförts på flera håll. En firma har lyckats konstruera en engångssond, som i kombination med en analysator visat god funktion. Figur 36:d visar principen för detta system, som går under namnet HYDRIS. Mätsondens ände, som doppas i smältan, består av en porös keramisk kopp genom vilken kvävgas spolas. Sonden kopplas till analysatorn via en slang och genom denna pumpas kvävgas, som får cirkulera genom analysatorn.
Då kvävgasen bubblar genom smältan sker en upptagning av väte från stålet och detta fortgår tills partialtrycket av väte i kvävgasen är samma som partialtrycket av väte i stålsmältan. Jämvikten bestäms av Siverts lag,

där (K) är en temperaturkonstant, (f) en korrektionsfaktor för stålsammansättningen samt (PH2) vätets partialtryck i bar. Mättiden är cirka 1 minut från det sonden doppats i smältan, varefter vätehalten kan avläsas på en display.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}